|
 ¿Qué
es la enfermedad de Charcot-Marie-Tooth?.
Características de CMT
¿Qué causa la CMT?
¿Cómo se hereda?
Síntomas y signos
Clínica
Biopsia
¿Es contagiosa
la enfermedad Charcot-Marie-Tooth?
¿Hay algún
tratamiento o curación para esta
enfermedad?
Drogas
contraindicadas. (según CMT 1Canada)
Ortopedia
Electrofisiología ¿Qué
es la enfermedad de Charcot-Marie-Tooth?.
Características de CMT
¿Qué causa la CMT?
¿Cómo se hereda?
Síntomas y signos
Clínica
Biopsia
¿Es contagiosa
la enfermedad Charcot-Marie-Tooth?
¿Hay algún
tratamiento o curación para esta
enfermedad?
Drogas
contraindicadas. (según CMT 1Canada)
Ortopedia
Electrofisiología |
|
|
|
1 |
|
1- ¿Qué es la enfermedad de
Charcot-Marie-Tooth?. |
|
|
|
La enfermedad de Charcot-Marie-Tooth
o atrofia muscular peronea es un
trastorno hereditario relativamente
frecuente del sistema nervioso
periférico (por lo común de herencia
autosómica dominante), |
|
|
|
 |
|
|
|
|
2 |
|
2- Características
de CMT. |
|
|
|
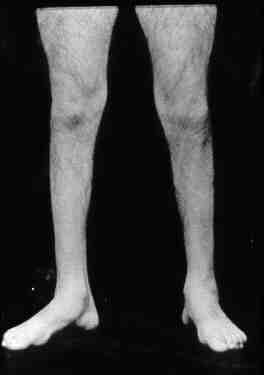
Se caracteriza
por debilidad y atrofia,
principalmente en los músculos
distales de la pierna peroneos,
tibial anterior, gemelos (foto
1 y 2). Se
presenta con déficit preferentemente
motor y en menor grado sensitivo de
distribución distal y carácter
progresivo, y con un comienzo en los
miembros inferiores y eventual
posterior afectación de los
superiores. |
|
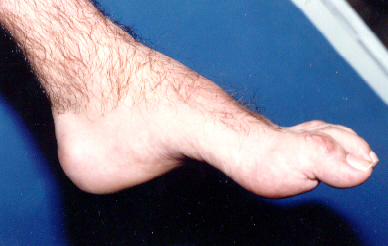
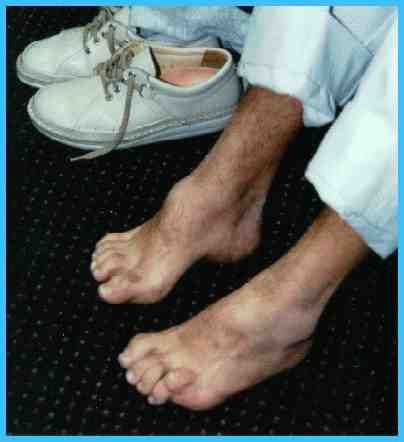
El pie
arqueado es una de las primeras
muestras de este desorden. Mientras
que progresa la enfermedad, las
deformidades estructurales del pie
ocurren. El paciente desarrolla un
pie cavus en forma de punta de
martillo (foto
3). Los
esguinces del tobillo son
manifestaciones frecuentes. La
pérdida progresiva del músculo
conduce a dificultad al correr y el
balance. Para evitar tropezar los
pacientes levantan sus rodillas
inusualmente hacia arriba dando por
resultado alto paso del steppage. En
algunos pacientes, la debilidad del
músculo puede también ocurrir en los
muslos. El pie plano se considera
también en pacientes con CMT. |
|
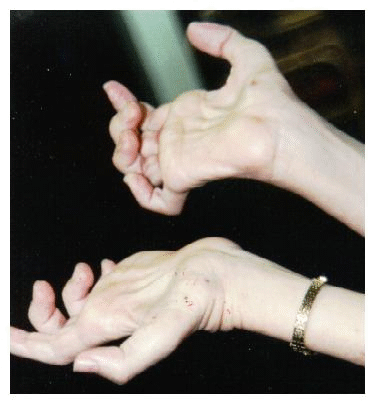
La función de
la mano también llega a ser afectada
debido a la atrofia progresiva (foto
4),
haciendo muy difícil la escritura y
las actividades manuales. |
|
La pérdida de función del nervio en
las extremidades también conduce a
la pérdida sensorial. Se disminuye
la capacidad de distinguir caliente
y frío también el sentido del tacto. |
|
Los músculos en las extremidades se
debilitan debido a la pérdida de
estímulo por los nervios afectados. |
|
Además CMT es un desorden en el cual
el defecto está en los nervios que
controlan los músculos no en estos.
La debilidad muscular ocurre como
consecuencia de la afectación del
nervio |
|
F |
|
Foto 1 y 2 |
 |
 |
| |
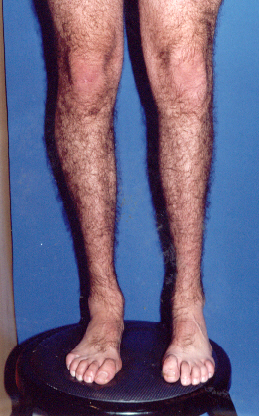 |
Uno de nuestros pacientes portador
de CMT2, aunque en un estado
avanzado de atrofia muscular se ha
logrado que mejore el trofismo
muscular a nivel de músculos
gemelos, usa aditamento tensor
-corrector- para caída de pie (con
plantilla para sostén). |
|
f3
volverr |
|
Foto 3 |
|
 |
|
f4volver |
|
Foto 4 |
|
 |
|
|
|
|
|
|
|
|
3 |
|
3- ¿Qué causa la CMT?. |
|
|
|
CMT es causado por defectos en genes
que afectan el funcionamiento de los
nervios periféricos. Los genes
proveen las instrucciones o mapas
para las proteínas en el cuerpo.
Cuando un gen es anormal, la
proteína también es anormal o
alterada. Los genes que estén
alterados causan CMT y afectan ya
sea a la fibra de los nervios o a
las capas de mielina que los rodea. |
|
Se han identificado tres genes que
cuando son anormales resultan en el
tipo 1 de CMT. Estos genes están en
los cromosomas 1, 17 y X. Todos
afectan la mielina y el tipo del
cromosoma X puede afectar el nervio
en sí. |
|
|
|
|
|
|
|
|
4 |
|
4- ¿Cómo se hereda?. |
|
|
|
CMT se hereda generalmente en un
modelo dominante autosomal. Esto
significa que si un padre tiene la
enfermedad (el padre o la madre)
pasaría en un 50% a cada niño. |
|
CMT se puede también heredar en
forma recesivo o un modelo
X-x-linked. Para determinar el
modelo de la herencia, cada paciente
de CMT debe consultar al consejero
genético, al neurólogo o a otro
médico con conocimientos de la
enfermedad. |
|
|
|
|
|
|
|
|
5 |
|
5- Síntomas y signos. |
|
|
|
Los síntomas de CMT varían
grandemente en cada individuo.
Ningún médico puede predecir como
usted será en 10 o 20 años y si
alguno lo hace no debe ser tenido en
cuenta. Cómo se manifiesta su CMT
depende de su forma de vida, las
actividades, las tensiones, dieta y
todo que hace en su vida. Puesto que
dos personas no son iguales cada CMT
es diferente. Hay personas
portadores de la enfermedad de CMT
de edad avanzada que llevan una vida
activa sin contratiempos. De ahí que
muchas personas portadores de CMT no
sepan que tienen esta enfermedad. |
|
Los pacientes de tipo 1 presentan en
la mitad de la infancia un pie caído
y una atrofia muscular distal
lentamente progresiva que produce
una "deformidad en pierna de
cigüeña". Más tarde se inicia una
atrofia de la musculatura intrínseca
de la mano. Hay una disminución de
la sensibilidad vibratoria, dolorosa
y térmica, con distribución en
"guantes y calcetines". Los reflejos
tendinosos profundos están abolidos.
Las deformidades de los pies, con
arcos pedios altos o dedos en
martillo, pueden ser las únicas
manifestaciones en los individuos de
la familia más afectados, portadores
del rasgo. Los pacientes de tipo 1
tienen velocidades de conducción
nerviosa lentas, con latencias
distales prolongadas. Las muestras
anatomopatológicas ponen de
manifiesto una desmielinización y
una remielinización segmentarias.
Pueden palparse nervios periféricos
agrandados. La enfermedad progresa
lentamente y es compatible con una
duración de vida normal.
Clínicamente, los pacientes tipo 2
suelen presentar debilidad en una
fase más tardía de la vida, y la
evolución del proceso es más lenta.
Las velocidades de conducción
nerviosa son relativamente normales,
pero los potenciales evocados son de
baja amplitud y las biopsias ponen
de manifiesto una degeneración
Walleriana. |
|
La neuropatía intersticial
hipertrófica (enfermedad de Dejerine-Sottas),
llamada CMT 3 es un trastorno
autosómico recesivo poco frecuente
de los nervios periféricos, que
aparece en la infancia con una
debilidad progresiva y pérdida de la
sensibilidad, con abolición de los
reflejos tendinosos profundos.
Inicialmente se asemeja a la
enfermedad de Charcot-Marie-Tooth,
pero la debilidad motora progresa
con mayor rapidez. Se trata también
de un trastorno de desmielinización
remielinización, con agrandamiento
de los nervios periféricos y "bulbos
de cebolla" en la biopsia. |
|
Alguna personas con CMT también
experimenta problemas con la
propiocepción que no es otra cosa
que la capacidad de decir donde está
su cuerpo en el espacio alrededor de
él. |
|
Con un diagnóstico temprano y
tomando el cuidado necesario, la
mayoría de las personas con CMT
vivirá una vida normal sin demasiada
dificultad, aunque no hay que negar
el hecho de que algunas personas
tienen problemas severos. |
|
La esperanza de vida no se acorta. |
|
|
|
|
|
|
|
|
6 |
|
6- Clínica. |
|
|
|
Atendiendo solo a las
características clínicas, es
imposible determinar la pertenencia
de un paciente individual a las
formas CMT1 o CMT2. Sin embargo, se
han sugerido ciertas diferencias
clínicas entre los grupos (Harding y
Thomas, 1980b). Así, mientras que
CMT1 tiene su edad de presentación
en las dos primeras décadas, el
rango de edades de comienzo de CMT2
es muy variable y se sitúa entre la
segunda y la séptima décadas de la
vida. Para un grado equivalente de
debilidad muscular, la atrofia
muscular parece ser más marcada en
CMT2. En CMT2 los músculos de la
pantorrilla sufren una afectación de
igual intensidad a la de los del
compartimiento anterolateral de la
pierna, y no es raro encontrar clara
asimetría en su distribución
(Berciano et al., 1986). Por el
contrario, la afectación de la
musculatura intrínseca de manos, el
grado de arreflexia , la pérdida de
la sensibilidad y la deformidad de
los pies en cavo parecen ser más
intensos en CMT1. Las raras formas
autosómico recesivas (Harding y
Thomas, 1980a; Ouvrier et al., 1981)
son siempre más graves que las más
habituales formas dominantes.
Comienzan muy precozmente con
retraso en la deambulación y
tropiezos y caídas frecuentes. Muy
pronto se aprecia un déficit motor
distal en miembros inferiores y
también en manos. La debilidad en
manos es más pronunciada que en las
formas dominantes. La presencia de
dolor u otros síntomas sensitivos es
rara, pero la exploración de la
sensibilidad suele mostrar pérdida
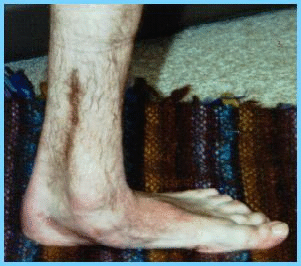
de la propiocepción. El pie cavo no
es constante (foto 5). |
|
f5 |
|
Foto 5. |
 |
| |
|
|
|
|
|
|
7 |
|
7- Biopsia. |
|
|
|
La introducción en los años 60 y 70
de los estudios de conducción
nerviosa y la práctica de biopsia de
nervio periférico permitieron
establecer dos tipos principales de
CMT (Dyck y Lambert, 1968). Una la
forma hipertrófica, desmielinizante
o CMT tipo1, caracterizada por
descenso de las velocidades de
conducción nerviosa motora
periférica, desmielinización de las
fibras nerviosas mielínicas y
crecimiento hipertrófico de las
células de Schwann en "bulbos de
cebolla"; y la otra axonal o CMT
tipo 2, con velocidades de
conducción nerviosa periférica
normales o mínimamente descendidas y
atrofia axonal primaria con mínima
afectación de la mielina de las
fibras nerviosas. Aunque se han
encontrado con mayor frecuencia CMT1
que CMT2. |
|
|
|
|
|
|
|
|
8 |
|
8- ¿Es contagiosa la enfermedad
Charcot-Marie-Tooth?. |
|
|
|
No. Las enfermedades genéticas no
son contagiosas. |
|
|
|
|
|
|
|
|
9 |
|
9- ¿Hay algún tratamiento o curación
para esta enfermedad?. |
|
|
|
No en estos momentos no hay ninguna
curación para CMT |
|
Por ahora el tratamiento incluye
terapia física, sostenes para la
parte inferior de las piernas,
ortesis de tobillo, correctores para
la caída del pie o plantillas en los
zapatos y a veces cirugía para
corregir las deformidades. |
|
|
|
|
|
|
|
|
10 |
|
10- Drogas contraindicadas. (según
CMTI de Canadá). |
|
|
|
Adriamycin - para el cáncer.
Alcohol.
Amiodarone - para el latido
irregular del corazón.
Cloranfenicol - antibiótico.
Cis-platino - para el cáncer.
Dapsone - para enfermedades raras
crónicas y ciertas de la piel,
también para la lepra.
Diphenylhydantoin ( Dilantin
) - para los asimientos y el dolor.
Disulfiram ( Antabuse ) -
alcohólicos.
Ethionamide - para la tuberculosis.
Flagyl ( Metronidazole ) -
para la infección de los trichomonas
- lea las escrituras de la etiqueta
en las prescripciones para las
infecciones bacterianas vaginales,
puede ser encontrado conjuntamente
con otra droga para las infecciones
vaginales, también se utilizó como
tópico para el acné y oral como
antibiótico de algunos dentistas y
doctores.
Glutethimide ( Doriden ) -
píldora para dormir.
Hydralazine ( Apresoline ) -
tensión arterial alta.
Isoniazid ( INH ) - para la
tuberculosis.
Misomidazole - para el cáncer.
Nitrofurantoin ( Furadantin,
Macrobid, Macrodantin ) - para
la infección de la zona urinaria.
Óxido nitroso (inhalación relanzada
crónica) - un anestésico.
Penicillamine - artritis
reumatoidea.
Perhexiline ( Pexid ) - para
la angina.
Pyridoxine ( vitamina B6 ) -
una vitamina.
Suramin, Taxol, Vincristine - para
el cáncer.
Tabaco.
Megadosis de las vitaminas A, B6 y D
puede ser dañoso. Controle
cuidadosamente con su doctor y
farmacéutico antes de tomar
cualquier droga. Pida que busquen
las palabras " podría causar la
neuropatía periférica " en la
descripción de la droga. En casi
todas las condiciones en las cuales
estas drogas se utilizan puede ser
encontrado una alternativa . |
|
Si usted necesita litio, o el
misomidazole para el cáncer u otras
drogas del cáncer, discuta las
repercusiones posibles con su
doctor. |
|
Zoloft puede causar fatiga pero no
es necesariamente peligroso a los
que lo necesiten. |
|
Recuerde que todas las drogas tienen
efectos secundarios. Usted podría
ser extremadamente sensible a las
drogas no enumeradas aquí. |
|
|
|
|
|
|
|
|
11 |
|
11- Ortopedia. |
|
|
|
La cirugía puede ayudar a problemas
del pie, del tobillo, de la mano,
del dedo, de la espina dorsal y de
la cadera. las correcciones del
tobillo-pie puede también ayudar a
una persona a correr sin tener
caídas, los correctores para caída
del pie en el zapato o en una
plantilla sostenida a la
pantorrilla ayudan a aliviar el
dolor experimentado y a correr y le
da a la persona un paso
estéticamente mas normal y mejorado. |
|
|
|
Procedimientos sugeridos para
personas con CMT. |
|
|
|
Procedimientos del tendón: |
|
alargar el tendón de Aquiles |
|
transferencia tibial anterior del
tendón |
|
procedimientos del clawtoe
(Girdlestone-Taylor , Jones).ambos
para la caída del pie. |
|
|
|
Procedimiento para ser realizado: |
|
artrodesis triple |
|
alargar el tendón de Aquiles. |
|
fasciotomía plantar |
|
|
|
Otro: |
|
Cirugía de la espina dorsal para
corregir la escoliosis. |
|
|
|
|
|
|
|
|
12 |
|
12- Electrofisiología. |
|
|
|
El criterio fundamental que ha
permitido la división de CMT en
tipos 1 y 2 es el valor divisorio de
la velocidad de conducción nerviosa
motora (VCM) situado en 38 m/s (Dyck
y Lambert, 1968; Harding y Thomas,
1980b). En todos los miembros
afectos de familias con CMT2 existe
una perfecta concordancia en sus
valores de VCM que son 38 m/s. Como
es lo característico de una
axonopatía distal, la normalidad o
mínimo enlentecimiento de la VCM
suele acompañarse de una caída en la
amplitud de los potenciales motores
en miembros inferiores, pero no
siempre ocurre así en miembros
superiores. Al estimular los nervios
peroneal o mediano los potenciales
motores solo están ausentes en
aquellos casos con atrofia muscular
marcada, y los músculos
correspondientes muestran
denervación y reinervación crónica
en el EMG. En cambio, en pacientes
asintomáticos las alteraciones
electrofisiológicas pueden ser
mínimas. Las alteraciones
encontradas en los potenciales
nerviosos sensitivos son
superponibles a las de los motores.
A diferencia de lo que ocurre con
CMT1 en que los trastornos
electrofisiológicos están
precozmente presentes y permiten un
rápido diagnóstico presintomático de
los individuos en riesgo, en CMT2
estas anomalías suelen establecerse
más tardíamente y progresan muy
lentamente a lo largo del tiempo, y
por ello el diagnóstico
electrofisiológico de portadores de
CMT2 puede ser difícil. Por otra
parte, las alteraciones
electrofisiológicas ya establecidas
no son específicas, y si la historia
familiar no es concluyente, pueden
conducir al diagnóstico erróneo de
neuropatía axonal adquirida
(diabética o alcohólica) (Teunissen
et al., 1997). Otro motivo de
confusión diagnóstica acontece en
familias sin transmisión varón-varón
y que corresponden a CMT1 ligada al
cromosoma X, al considerar como CMT2
a mujeres afectas o portadoras y que
característicamente tienen
alteraciones electrofisiológicas de
"tipo axonal" diferentes a las de
"tipo desmielinizante" encontradas
en los varones afectos (Nicholson y
Nash, 1993). |



